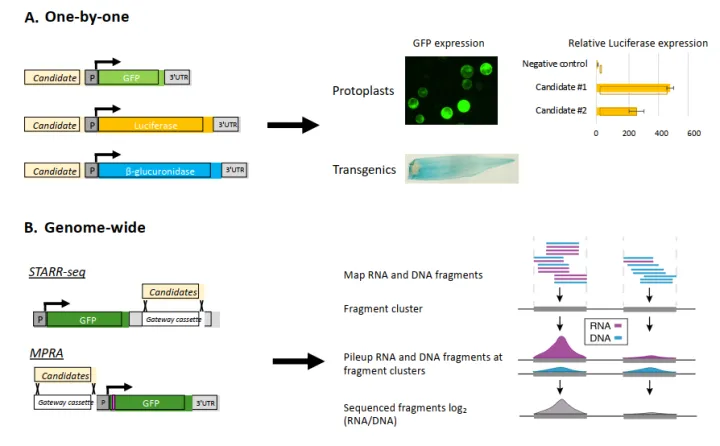
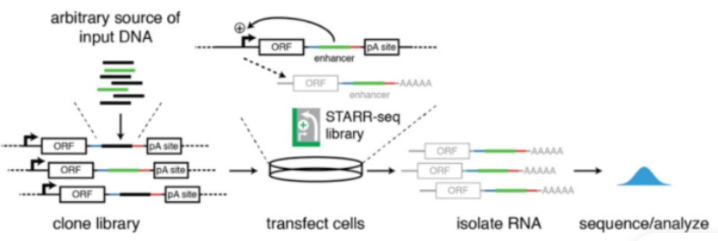
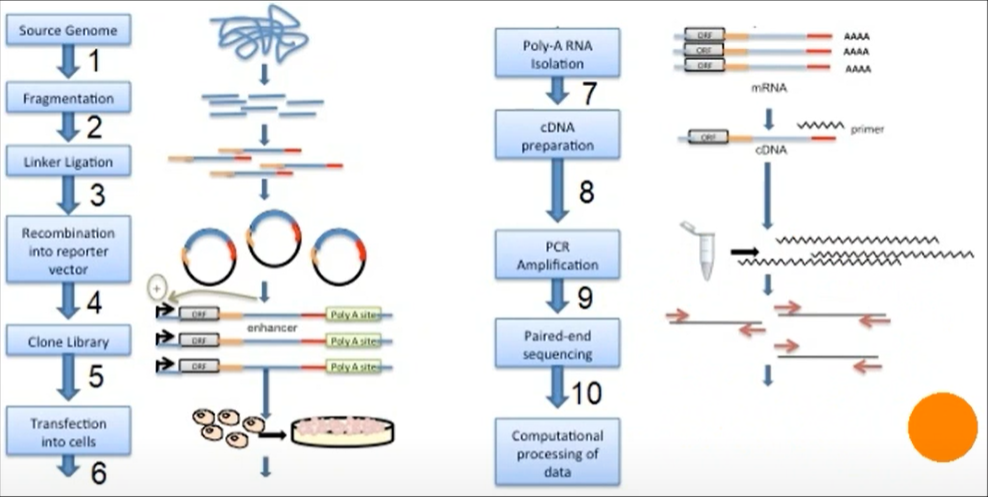
STARR-seq:该方法是用来评估启动子区域的增强子活性。
Epromoters: 指的是既可以作为promoters 同时可以作为远端基因的enhancers
enhancers和epromoters的鉴定流程如下图:
主要是从GEO数据库搜集目前已发表的
STARR-seq和MPRA数据,然后分别进行Enhancer peak calling ,鉴定具有enhancers和Epromoters活性的序列。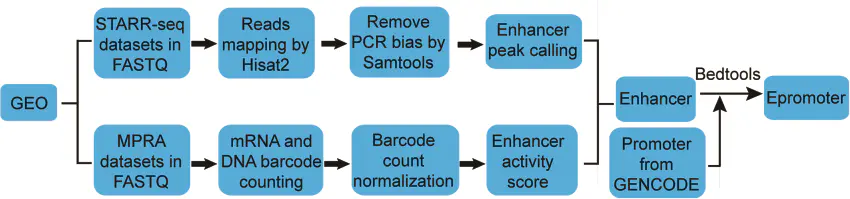
链接:https://www.jianshu.com/p/98ce9a3f3fe5
STARR-seq目前广泛应用于增强子活性检测。但传统的STARR-seq的准确性严重依赖于从报告基因reporter gene启动子开始的自转录mRNA的完全恢复。
在质粒构建过程中,polyadenylation site(PAS)被添加到报告基因的后端,由于这个是设计好的PAS用来给自转录self-transcripts (STs) 做聚腺苷酸化polyadenylation 的,称之为“DPAS”。但是,可能存在alternative另外的 polyadenylation site(PAS)在检测DNA序列中,也是受到了enhancer的潜在影响,称之为“APAS”。APAS在STARR-seq中是不会被检测到的。
链接:https://www.jianshu.com/p/57d18d57d34f
一种高通量的验证方法例如self-transcribing active regulatory region-sequencing(STARR-seq)能够对基因组范围内的CRMs进行大规模的评估。在STARR-seq中,基因组片段被打断并添加barcode,打断后的片段被克隆到报告基因的3’UTR区域,从而创建了一个报告基因库中衍生的RNA片段表明了CRMs的活性(图5B)。
https://zhuanlan.zhihu.com/p/440319953
https://news.sciencenet.cn/htmlpaper/2020/8/20208123422227357873.shtm
https://wenku.baidu.com/view/4ad853d4920ef12d2af90242a8956bec0975a5d4.html?_wkts_=1674140985734&bdQuery=starr-seq
https://www.science.org/doi/10.1126/science.1232542
Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq
SCIENCE17 Jan 2013Vol 339, Issue 6123pp. 1074-1077Abstract
Genomic enhancers are important regulators of gene expression, but their identification is a challenge, and methods depend on indirect measures of activity. We developed a method termed STARR-seq to directly and quantitatively assess enhancer activity for millions of candidates from arbitrary sources of DNA, which enables screens across entire genomes. When applied to the Drosophila genome, STARR-seq identifies thousands of cell type–specific enhancers across a broad continuum of strengths, links differential gene expression to differences in enhancer activity, and creates a genome-wide quantitative enhancer map. This map reveals the highly complex regulation of transcription, with several independent enhancers for both developmental regulators and ubiquitously expressed genes. STARR-seq can be used to identify and quantify enhancer activity in other eukaryotes, including humans.
Methodology[edit]
Genomic DNA is randomly sheared and broken down to small fragments. Adaptors are ligated to size-selected DNA fragments. Next, adaptor linked fragments are amplified and the PCR products are purified followed by placing candidate sequences downstream of a minimal promoter of screening vectors, giving them an opportunity to transcribe themselves. Candidate cells are then transfected with reporter library and cultured. Thereafter, total RNAs are extracted and poly-A RNAs isolated. Using reverse transcription method, cDNAs are produced, amplified and then candidate fragments are used for high-throughput paired end sequencing. Sequence reads are mapped to the reference genome and computational processing of data is carried out.[1]
https://www.sciencedirect.com/science/article/pii/S0888754315300100
https://en.wikipedia.org/wiki/STARR-seq
https://www.jianshu.com/p/98ce9a3f3fe5