病毒作为TME中的重要组分,在很多高分文章中已有论述:

Epstein-Barr virus (EBV)感染了大约90%的成年人口,尽管该病毒不会在大多数宿主中引发明显症状,但是EBV却具有很强的致癌能力,并被认为是多种恶性肿瘤的致病源,例如B细胞或NK-T细胞淋巴瘤,上皮癌,比如鼻咽癌(NPC)和胃癌(GC)等。除了具有肿瘤细胞转化特性外,EBV还可以影响肿瘤微环境(TME)中存在的细胞的特性和组成。比如,EBV在裂解和潜伏感染期间表达的特定蛋白质会导致CD4+、CD8+T细胞和自然杀伤(NK)细胞的激活,从而诱导抗病毒免疫。EBV如何影响TME参与到胃癌的具体过程仍有待于深入分析。

题目:鉴定 Epstein-Barr 病毒调节胃癌肿瘤免疫微环境的关键基因
杂志:Cell Proliferation
影响因子:IF=8.76
发表时间:2023年3月
研究背景
Epstein-Barr病毒(EBV)在感染正常细胞后参与胃癌(GC)的癌变,并诱导肿瘤微环境(TME)的组成高度可变。然而,目前仍然缺乏对与EBV免疫浸润调节相关的关键基因的系统生物信息学分析。因此,作者系统地分析了EBV感染与免疫浸润调节的关系,并筛选出关键基因构建预后模型。
数据来源
| 数据库/队列 | 数据库 | 数据类型 | 样本信息 |
| TCGA-STAD | TCGA | RNA-seq数据 | 265例EBV相关胃癌(GC)样本的基因表达及临床信息 |
| GSE66229 | GEO | RNA-seq数据 | 300例胃癌(GC)样本的基因表达及临床信息 |
| GSE51575 | GEO | RNA-seq数据 | 52例胃癌(GC)样本的基因表达及临床信息 |
研究思路
作者利用TCGA和GEO数据库中的胃癌(GC)相关数据基因表达和临床数据,来分析EBV感染与GC中免疫浸润概况之间的关联。应用加权基因共表达分析(WGCNA)来阐明与GC中EBV相关免疫浸润相关的基因功能模块。基于GC组织微阵列芯片,分析EBV-和EBV+肿瘤样本之间关键hub基因的表达,并利用单细胞测序数据,分析hub基因与免疫细胞浸润的相关性。最后基于临床样本进行hub基因的验证及功能探讨。
主要结果
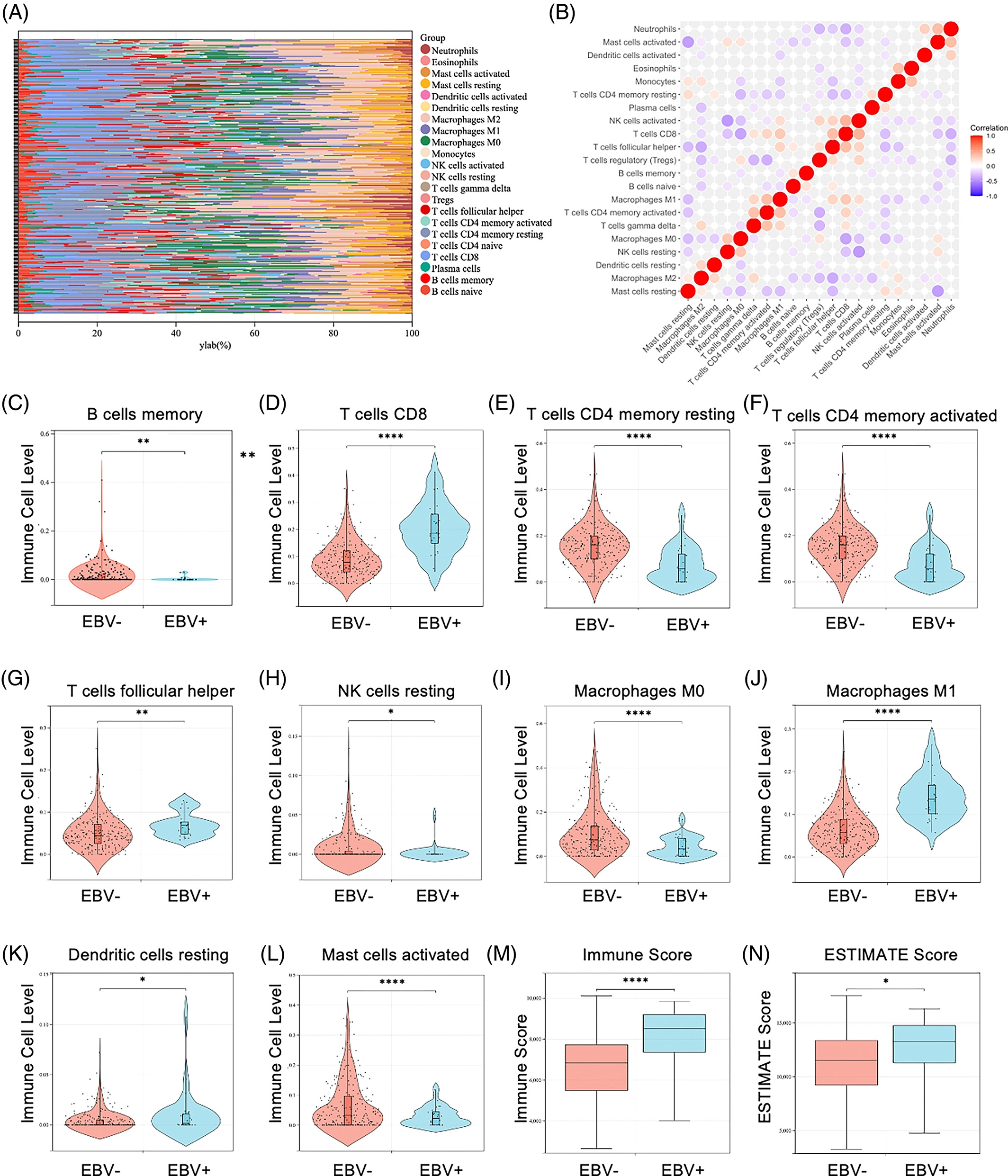
1. EBV感染改变了GC中免疫浸润的特征
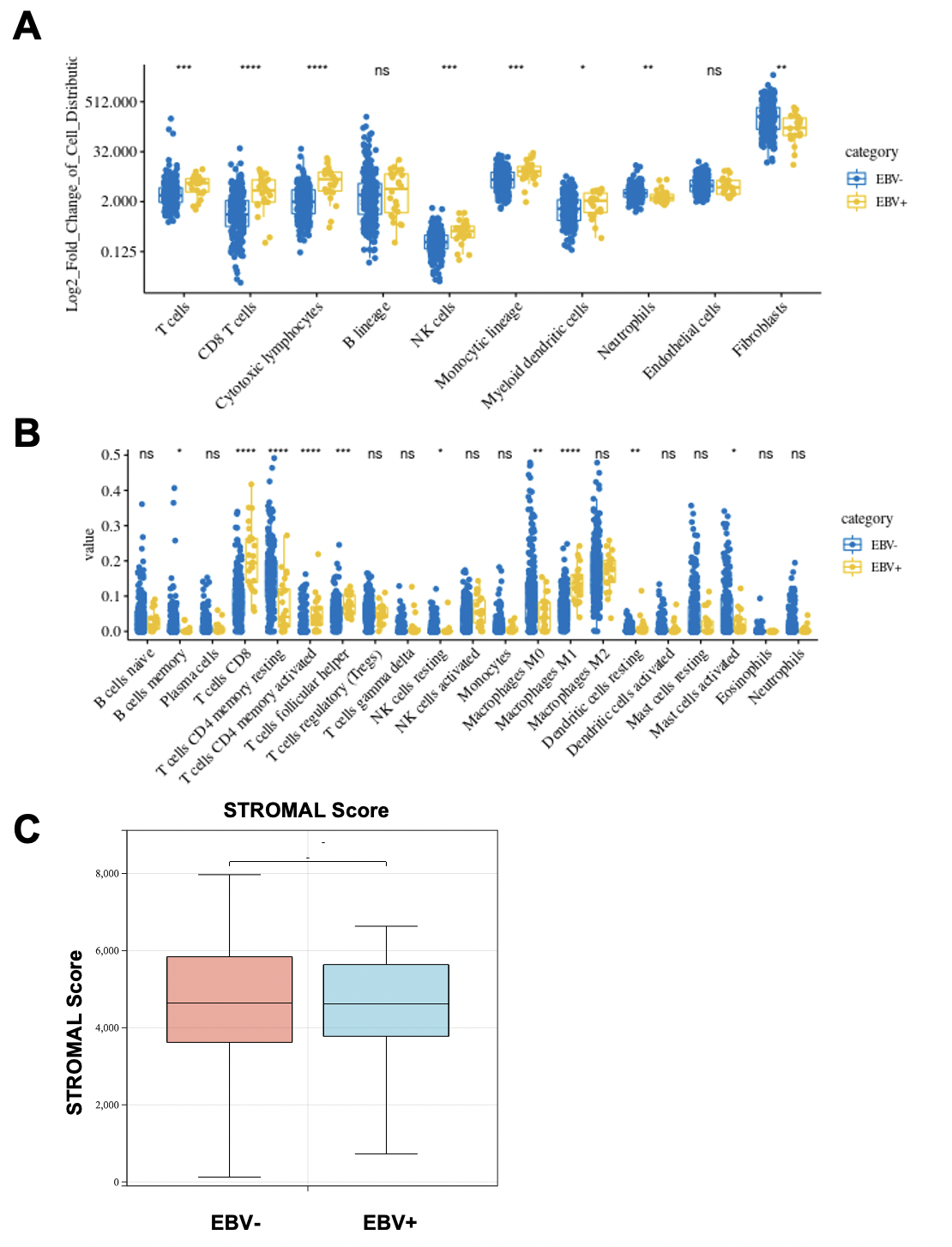
采用了CIBERSORT算法分析EBV感染是否影响GC中肿瘤浸润免疫细胞(TIIC)的比例,结果发现TIIC的分布因细胞种类而异,一些TIIC在EBVaGC肿瘤组织中有较多的免疫细胞,而另一些TIIC在EBV-肿瘤组织中的比例较低(图1A)。并且,EBV-和EBV+组织之间在数值上没有明显的相关性(图1B)。与EBV-样本相比,EBVaGC组织中CD8+T细胞(图1D)、T细胞滤泡辅助细胞(图1G)和巨噬细胞M1(图1J)的比例较高,而B细胞记忆(图1C)、T细胞CD4记忆静止(图1E)、NK细胞(图1H)、T细胞CD4记忆激活(图1F)、巨噬细胞M0(图1I)、树突状细胞(DC)静止(图1K)和肥大细胞激活(图1L)的比例较低。与EBV阴性组织相比,EBV感染显著增加了免疫功能(图1M)和评估评分(图1N),但对间质评分没有显著影响(图S1C)。这些结果表明EBV改变了免疫浸润的特征并促进了GC组织中的炎症反应。
图 1:
图 S1:
小结:
先看EBV感染是否影响了GC中肿瘤浸润免疫细胞(TIIC)的比例(肯定是有影响的,不然也不会写这篇文章了),以及大致的比例是个什么样的。然后看EBV感染的组织和EBV未感染的组织之中各肿瘤浸润免疫细胞有没有明显相关性,如果有相关性,说明一种免疫细胞的增加也会影响到另一种免疫细胞的增加,就要考虑共线性问题。这里EBV-和EBV+组织之间在数值上没有明显的相关性,就可以进行后续分析了。然后就是分析“每种细胞比例”、“免疫评分”、“总的ESTIMATE评分”在EBV-和EBV+组织中的高低是否有差异了。把有差异的细胞挑出来做小提琴图展示。
这一部分分三步走证明了EBV感染确实改变了GC中免疫浸润的特征,也找出了数个有差异的免疫浸润细胞的种类,且这些细胞之间没有共线性问题。
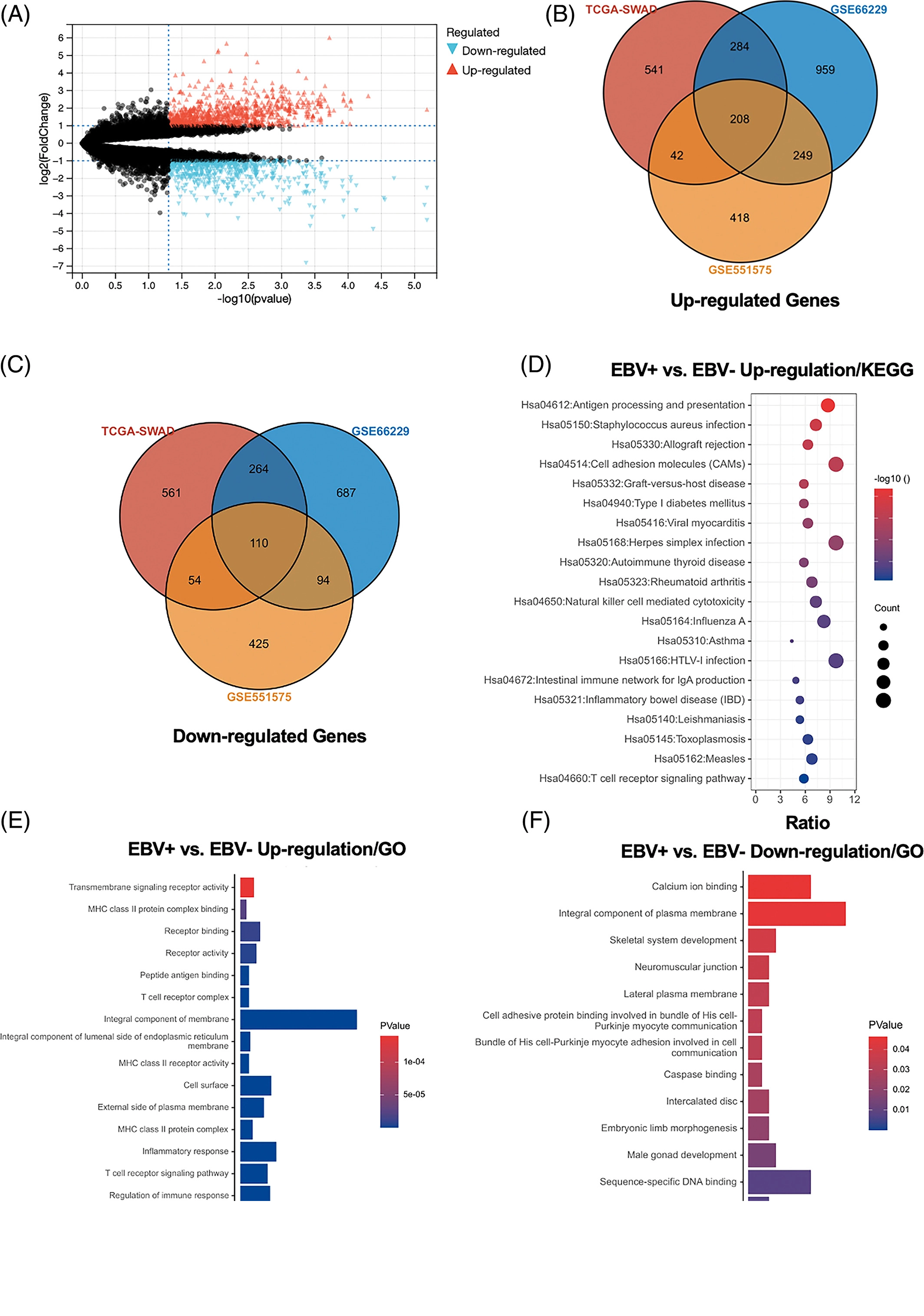
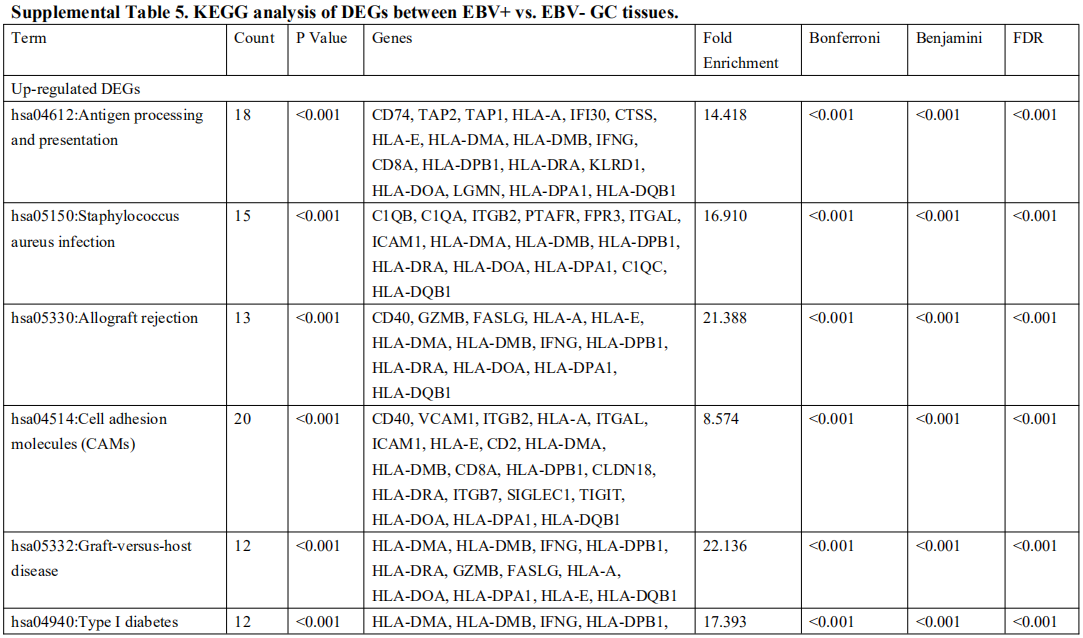
2. 三个基因集的DEG鉴定及功能分析
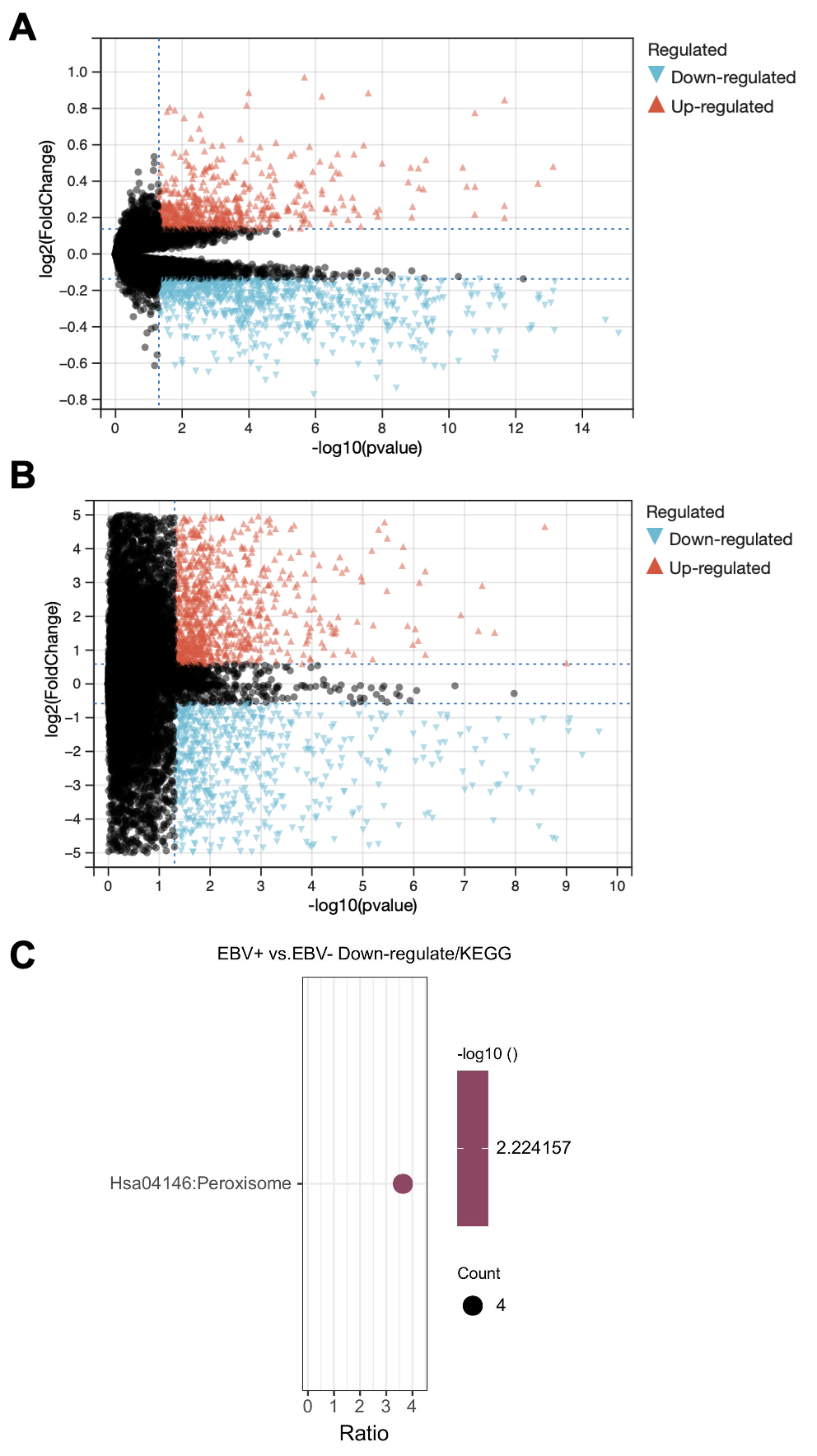
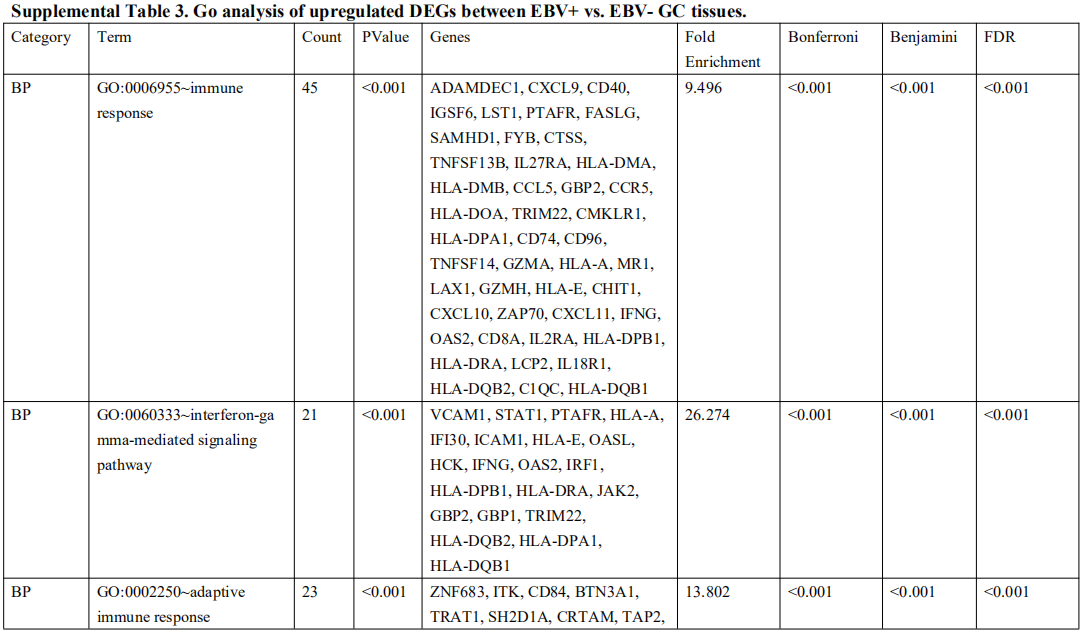
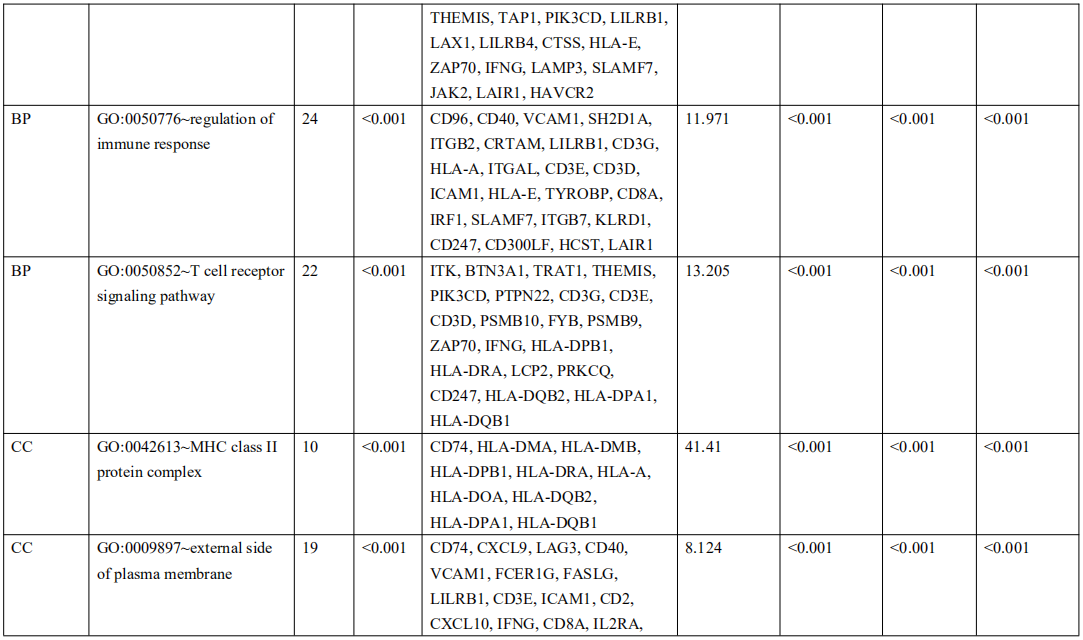
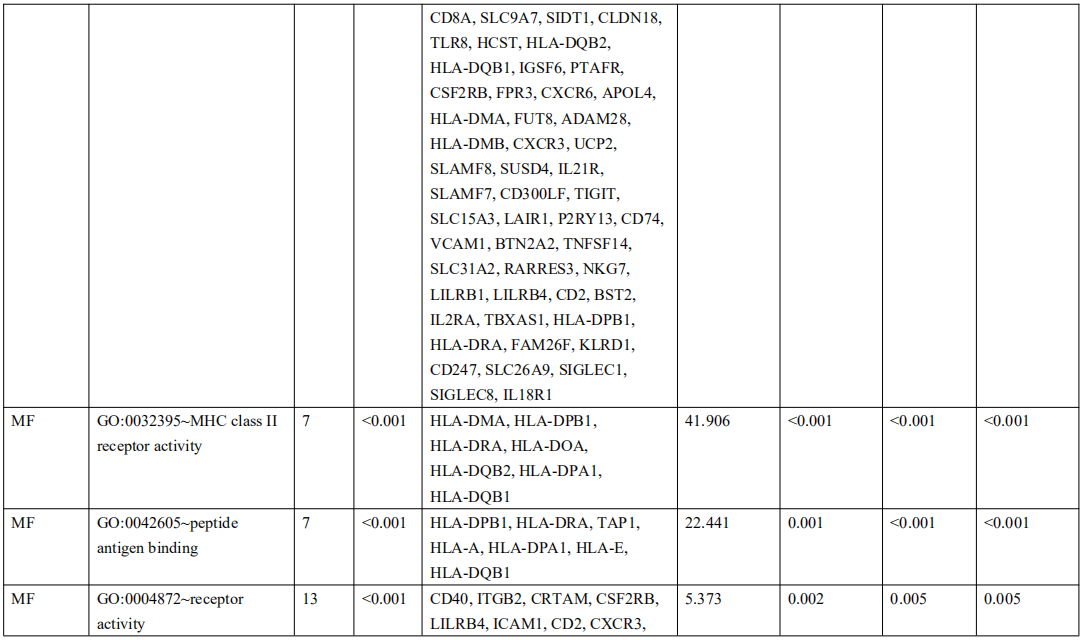
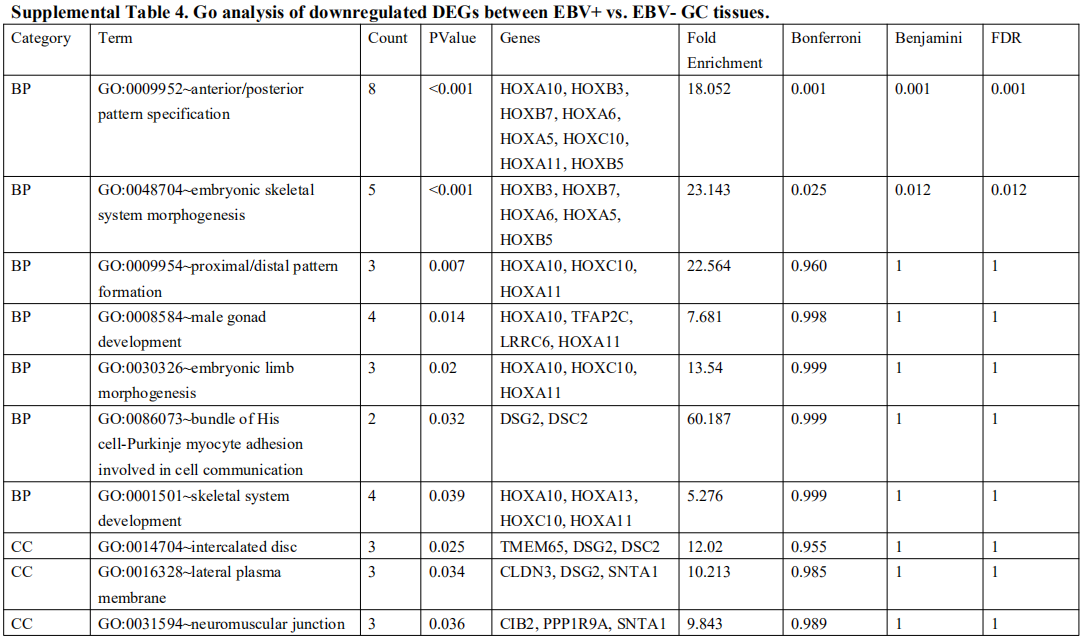
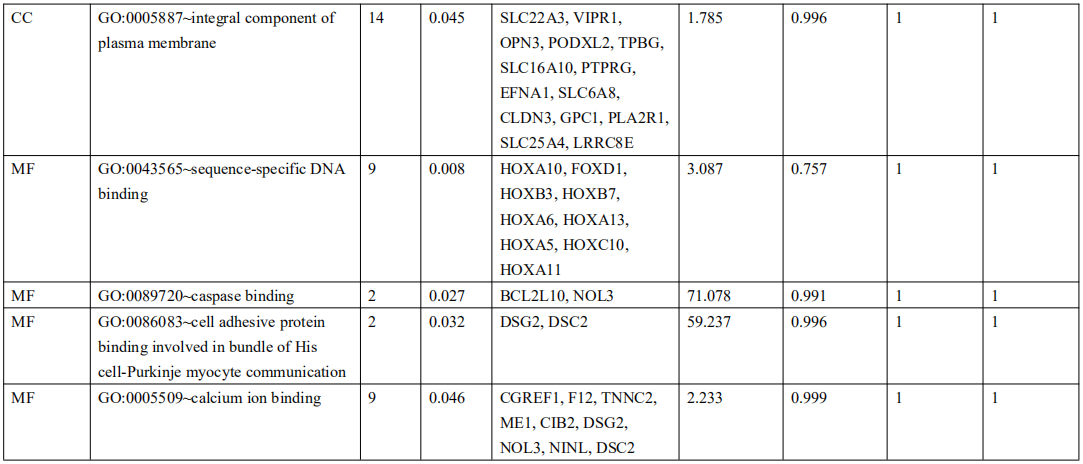
为了确定受EBV感染调控的差异基因,分析中包括了三个基因数据集。火山图显示了GSE551575(图2A)、GSE66229(图S2A)和TCGA-SWAD(图S2B)的DEG分布。这三个数据集包括208个相同的上调基因(图2B,表S1)和110个相同的下调基因(图2C和表S2)。然后,我们对上调或下调的DEGs进行了GO和KEGG途径的富集化分析。给出了生物过程、细胞化合物和分子功能以及15个KEGG信号通路的前五个重要的GO富集化结果。上调的GO分析包括免疫反应、干扰素介导的信号通路、适应性免疫反应、免疫反应的调节和T细胞受体信号通路等,它们与免疫和炎症反应显著相关(图2D和表S3)。下调的GO分析包括前后部模式规范、胚胎骨骼系统形态发生、近端/远端模式形成、男性性腺发育和胚胎肢体形态发生等。(图2E和表S4)。上调的DEGs主要集中在抗原加工和呈递、金黄色葡萄球菌感染、同种异体排斥反应、细胞黏附分子和移植物抗宿主病等方面(图2F和表S5)。然而,只有过氧化酶体途径与EBV+和EBV-GC组织之间的DEGs下调有关(图S2C和表S5)。这些结果提示,EBV感染与胃癌组织免疫反应的激活密切相关。
图 2:
图 S2:
表 S1:
表 S2:
表 S3:
表 S4:
表 S5:
小结:
对三个数据集进行了差异基因分析,logFC取1,p值取0.05,差异分析的结果用火山图的形式展示。然后三个数据集的差异基因取交集,得到208个上调差异基因和110个下调差异基因。最后对这些差异基因做GO和KEGG富集分析。
这一部分很简单,就是找了一堆差异基因,然后在GO和KEGG富集了一下。




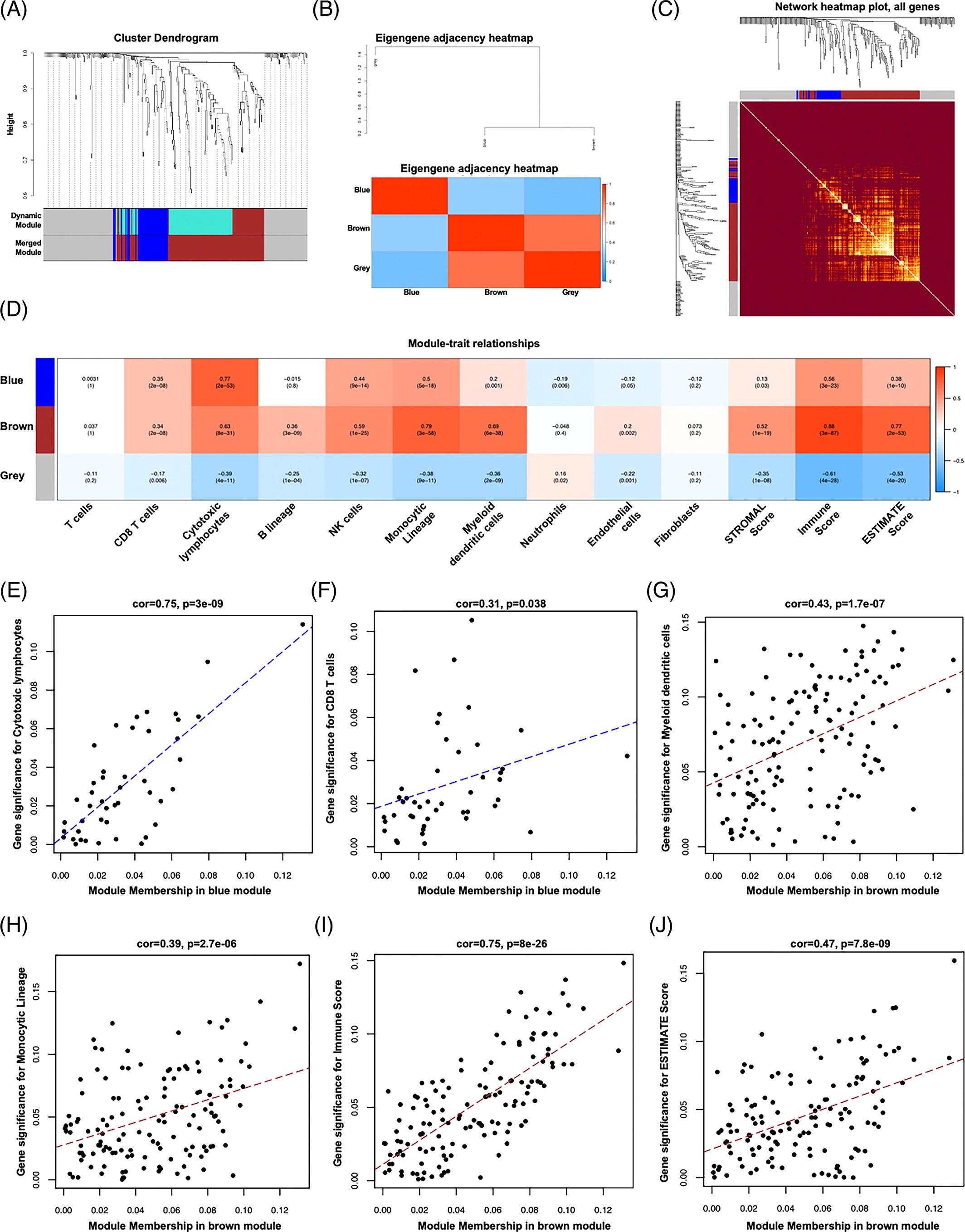
3. 参与免疫浸润反应基因模块的筛选
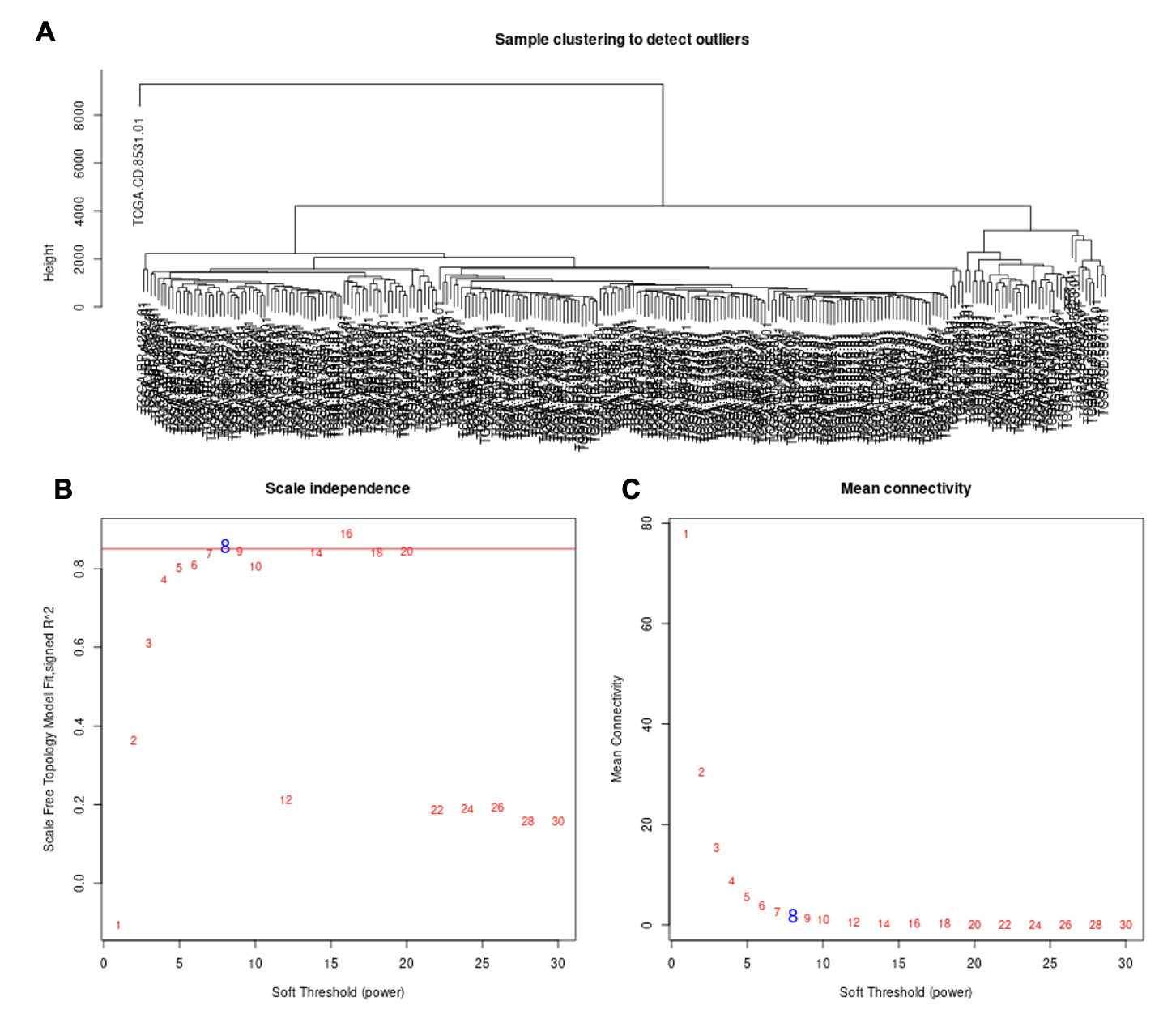
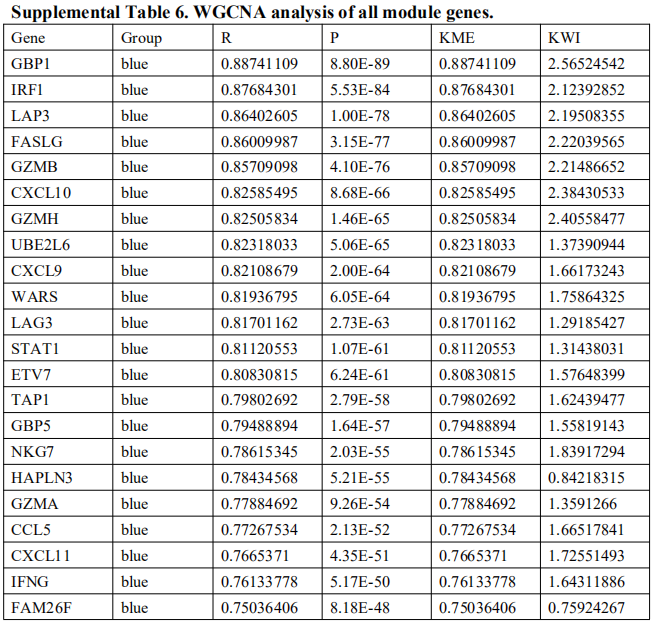
为了识别与免疫渗透相关的关键基因模块,使用EBV+和EBV-GC组织之间的所有DEG来构建层次聚类树。只发现了一个离群值样本(图S3A)。接下来,我们进行共表达分析,构建共表达网络。为了确保无标度网络,β的幂次被设置为软阈值(图S3B,C)。确定了三个共表达模块,分别以蓝色、棕色和灰色显示(图3A)。相关的热图如图3B所示。此外,热图的TOM值通过动态方法显示在模块划分的网络的基因表达中(图3C)。灰色模块显示低TOM,而棕色模块显示较高的TOM。为了进一步评估模块与免疫渗透的关联性,在聚类分析中使用了不同的免疫细胞(MCP算法)和ESTIMATE分数。如图3D所示,棕色模块与CD8 T细胞、细胞毒性淋巴细胞、B细胞系、NK细胞、单核细胞系、髓系树突状细胞、内皮细胞和所有ESTIMATE评分呈正相关。蓝模数与CD8 T细胞、细胞毒性淋巴细胞、NK细胞、单核细胞集落数、髓系树突状细胞及所有ESTIMATE评分均呈正相关。然而,灰色模块,这是未分配的基因,大多与免疫细胞和ESTIMATE分数呈负相关。所有模块基因如表S6所示。
4. GS与不同模块成员的关系
因为蓝色模块与细胞毒性淋巴细胞和CD8 T细胞有密切的模块-特性关系,而棕色模块在单核细胞集落、髓系树突状细胞、免疫评分和估计评分中更为明显。我们考察了它们之间的GS和MM。如图3E,F所示,蓝色模块显示明显的GS-MM,细胞毒性淋巴细胞(COR=0.75,p=3E-09)和CD8T细胞(COR=0.31,p=0.038)。此外,棕色模块与GS在单核细胞集落(Cor=0.43,p=1.7E-07,图3g)、髓系树突状细胞(Cor=0.39,p=2.7E-06,图3h)、免疫评分(Cor=0.75,p=8E-26,图3i)和估计评分(Cor=0.47,p=7.8E-09,图3J)中呈正相关。
图 3:
图 S3:
表 S6:
小结:
用WGCNA识别出了3个基因模块,然后就是WGANA分析的那一套流程,网上都能查到。
软阈值筛选、网络构建、层级聚类树展示各个模块(A)、绘制模块之间相关性图(B)、可视化基因网络 (TOM plot)(C)、关联表型数据(D),然后D中找几个感兴趣的关系画个点图。
WGCNA相关知识可参考:https://zhuanlan.zhihu.com/p/36132370,http://blog.genesino.com/2018/04/wgcna/,等。
5. EBVaGC中的免疫相关基因筛选及Hub基因鉴定
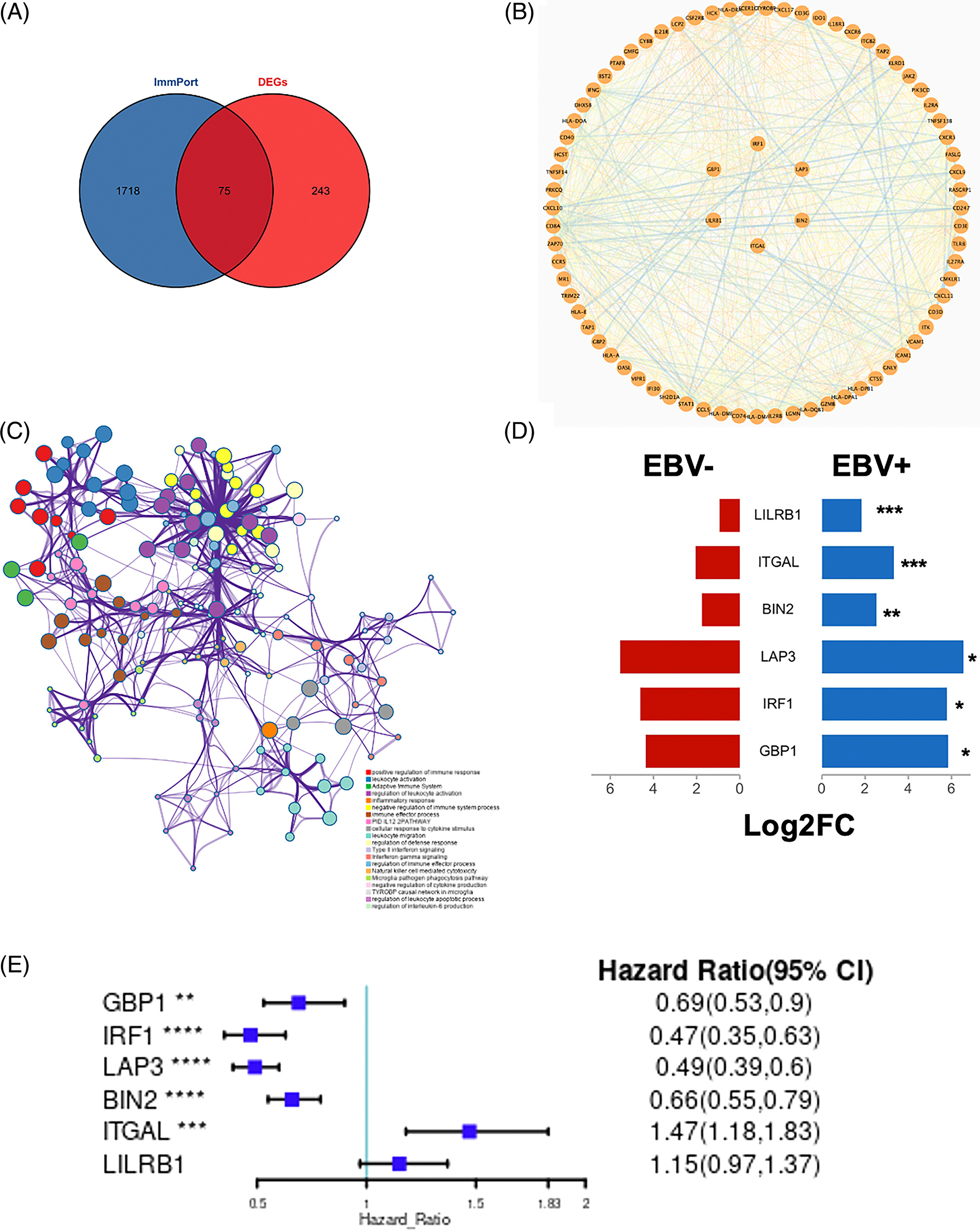
为了确定EBVaGC中的关键免疫相关基因,将DEGs与ImmPort数据库中的免疫相关基因列表进行交叉检索。如图4A所示,EBVaGC中共有75个基因被确定为关键免疫相关基因。PPI网络显示了这些基因之间的关系,内环的六个基因,包括干扰素调节因子1(IRF1)、亮氨酸氨基肽酶3(LAP3)、桥接整合子2(BIN2)、整合素亚单位αL(ITGAL)、白细胞免疫球蛋白样受体B1(LILRB1)和鸟氨酸结合蛋白1(GBP1)是蓝色和棕色模块的前三个hub基因(图4B)。
6. Hub基因鉴定
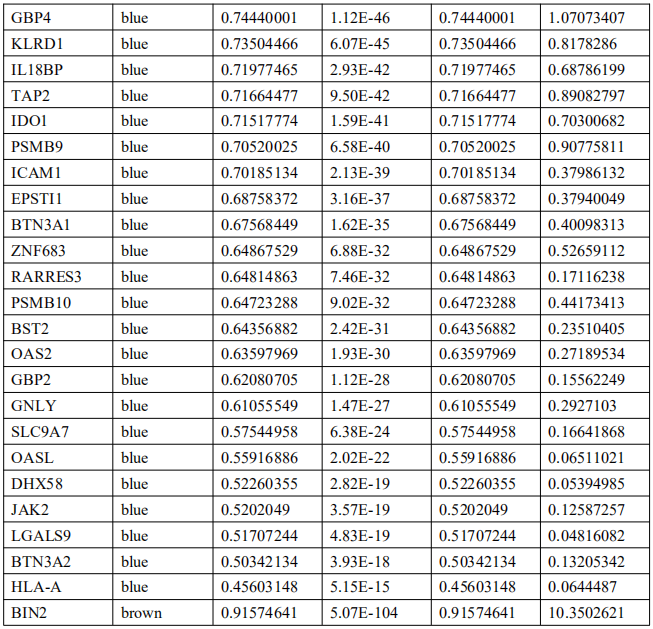
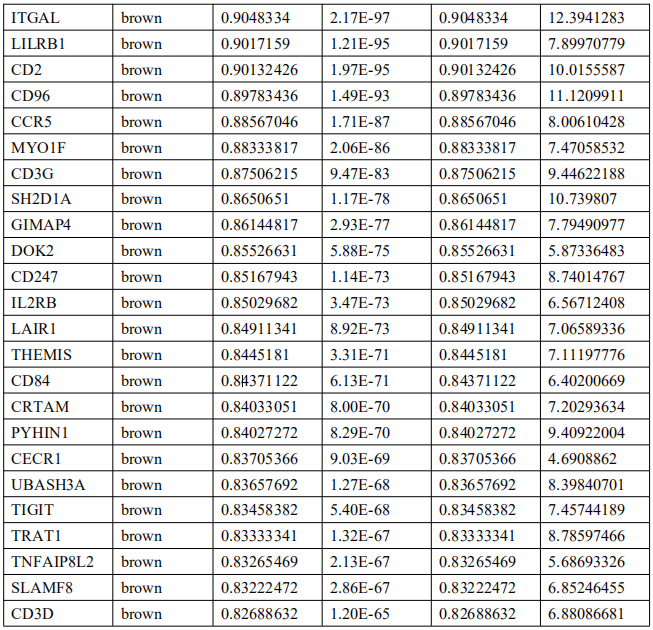
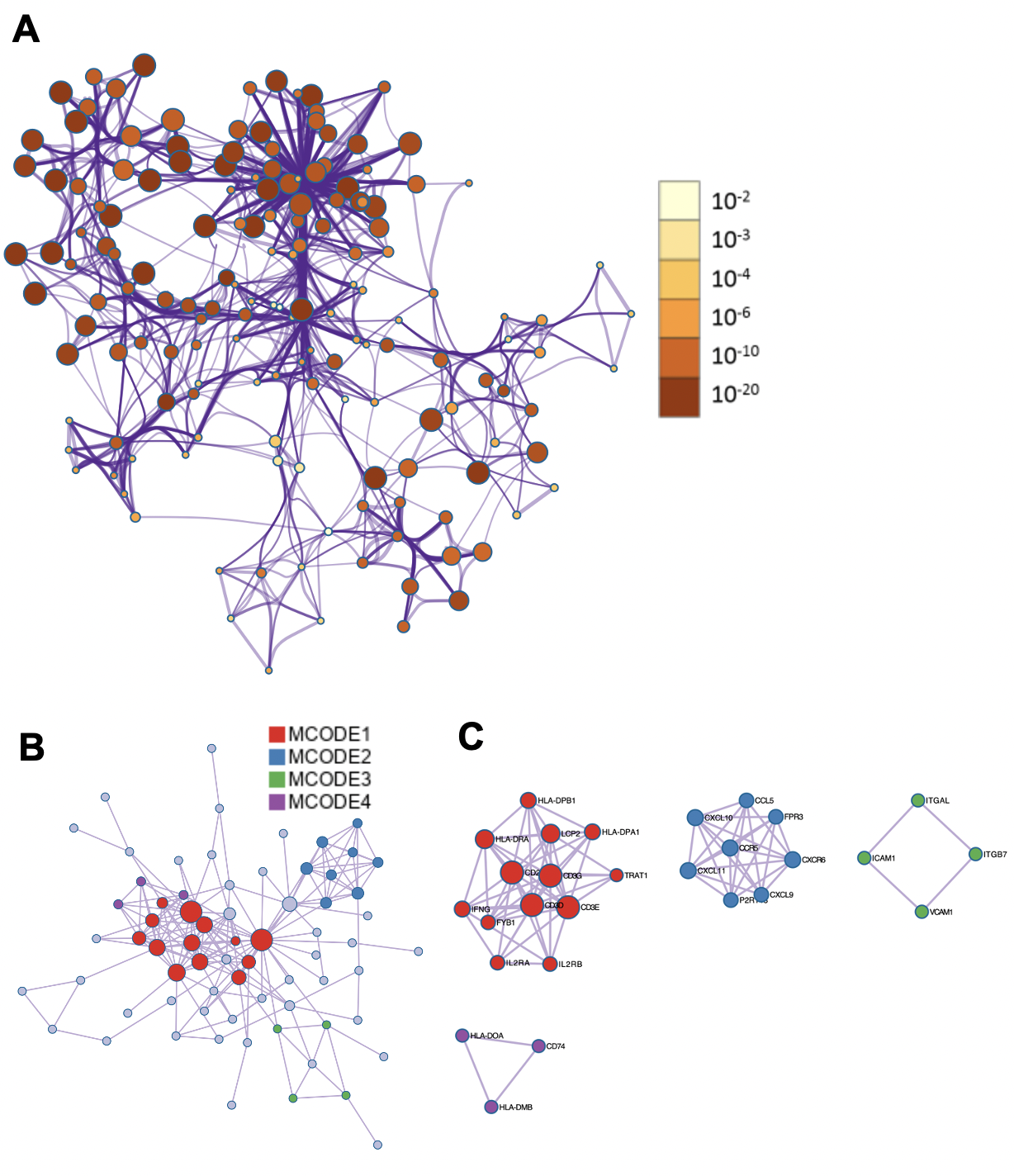
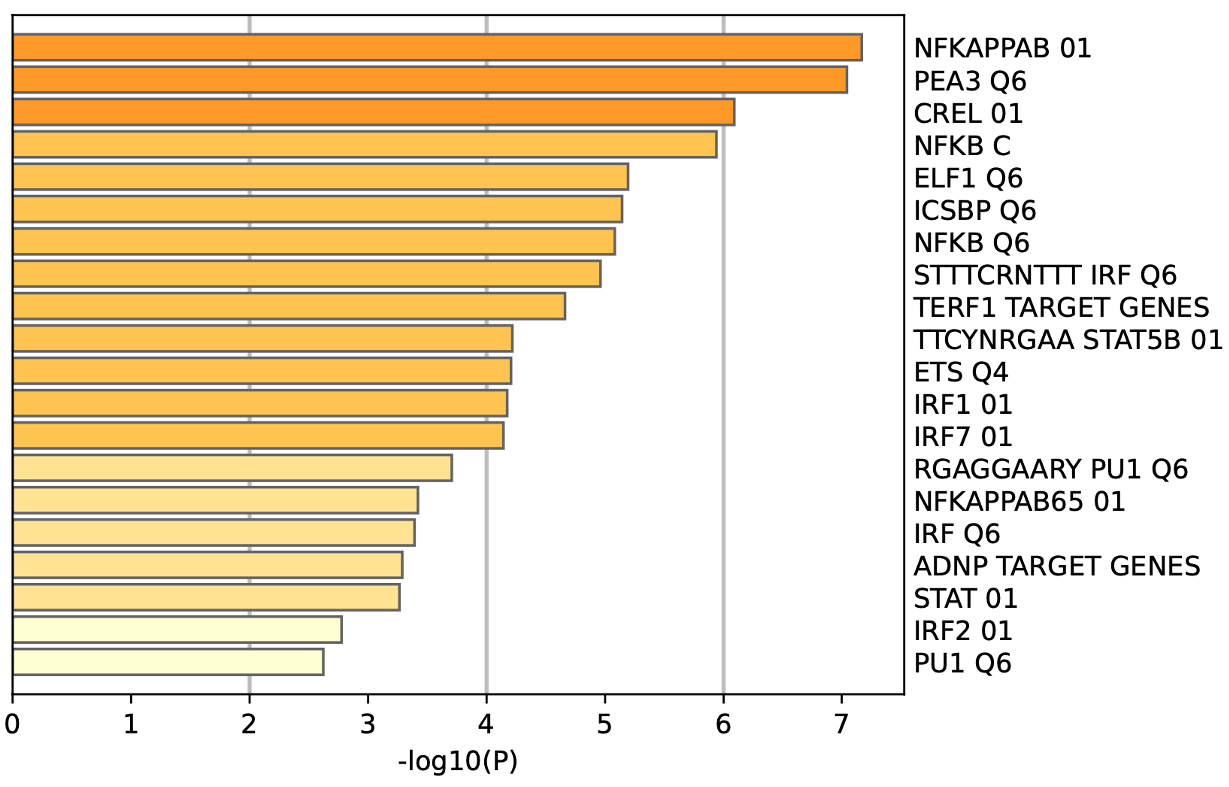
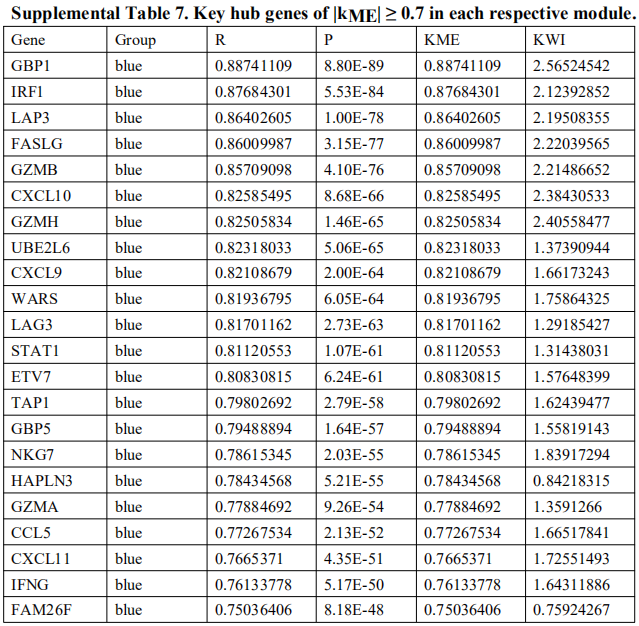
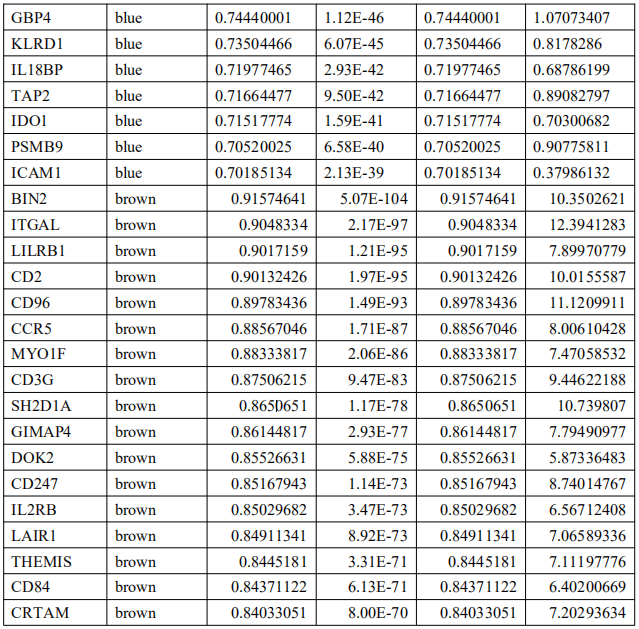
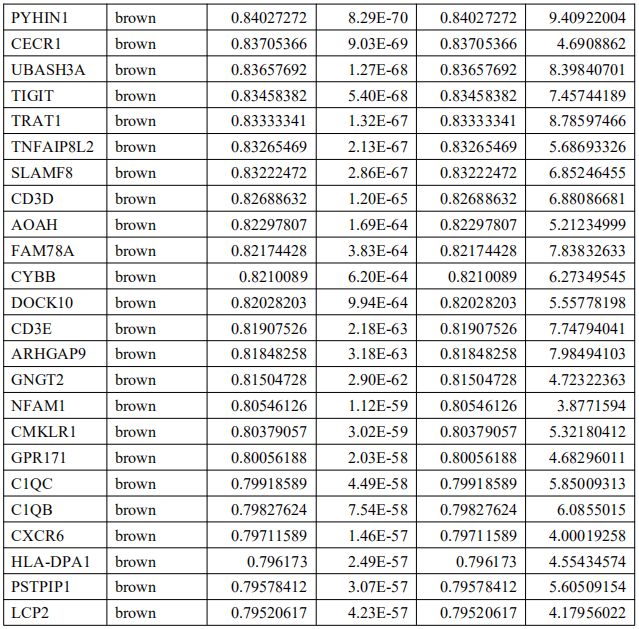
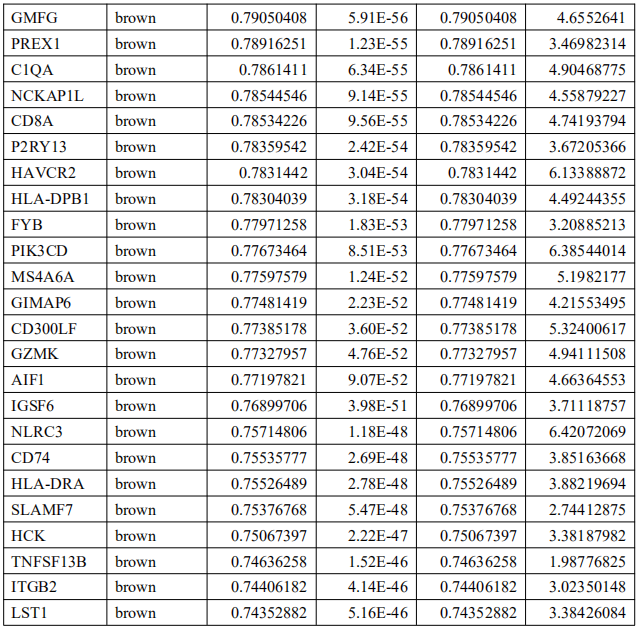
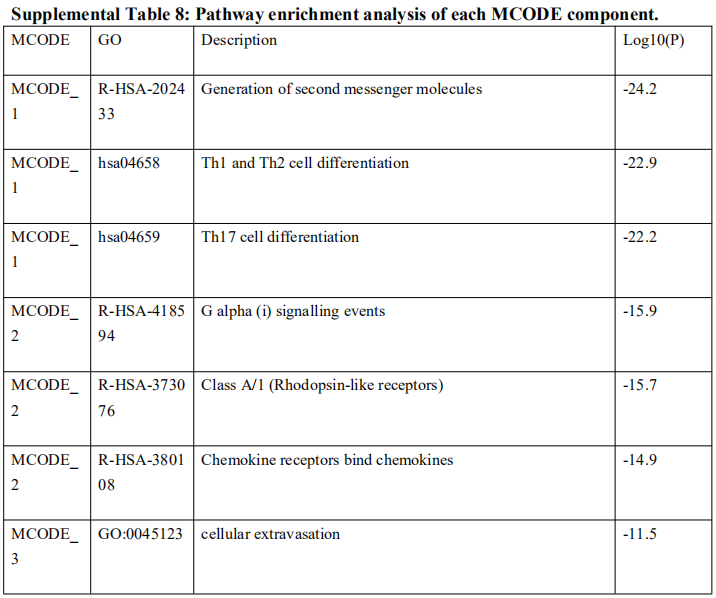
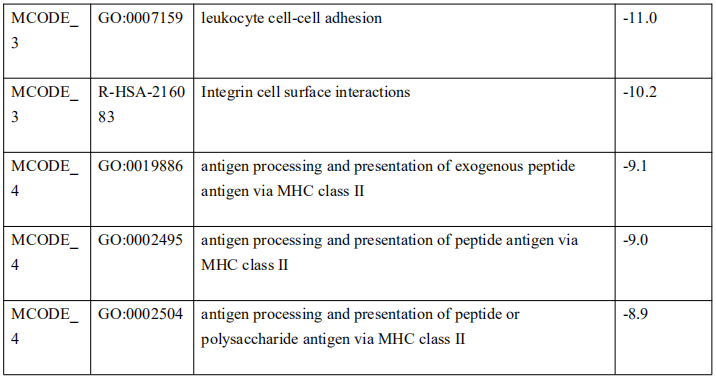
为了确定这些模块中的关键成分,|kME|≥0.7的基因被视为每个相应模块中的枢纽。如表S7所示,列出了所有|kME|≥0.7的Hub基因。蓝色和棕色模块分别包含29个和89个HUB基因。GO富集的子集已被选择并呈现为网络图。这些HUB基因在免疫反应、白细胞激活、适应性免疫系统、白细胞激活和炎症反应的调节等方面具有丰富的正向调节作用。(图4C和S4A)。然后,对物理相互作用进行了PPI富集化分析。MCODE算法已被应用于识别密集连接的网络组件。如图S4B所示,这些HUB基因被分为四个MCODE部分。路径和过程浓缩分析已独立应用于每个MCODE组件,并保留了按p值计算得分最高的三个术语,作为相应组件的功能描述,如图S4C和表S8中相应网络图下面的表格所示。为了分析哪些信号通路参与了这些HUN基因的调控,进行了KEGG分析。如图S5所示,前五个相关的信号通路是NFKAPPAB 01、PEA3 Q6、CREL 01、NFKB C和Elf1 Q6。选择BLUE模块的前三位HUB基因(GBP1[鸟苷结合蛋白1]、IRF1[干扰素调节因子1]、LAP3[亮氨酸氨基肽酶3])和Brown模块(BIN2[桥接整合子2]、ITGAL[整合素亚单位αL]、LILRB1[白细胞免疫球蛋白样受体B1])作为代表性基因进行下一步分析。
7. 关键HUB基因与GC的关系
如图4D所示,所有关键的HUB基因在EBVaGC样本中都过表达。有趣的是,如图4E所示,GBP1(HR=0.69,95%CI=0.53~0.9)、IRF1(HR=0.47,95%CI=0.35~0.63)、LAP3(HR=0.49,95%CI=0.39~0.6)和BIN2(HR=0.66,95%CI=0.55~0.79)的高表达提示GC患者预后良好。ITGAL高表达与预后不良相关(HR=1.47,95%CI=1.18~1.83)。
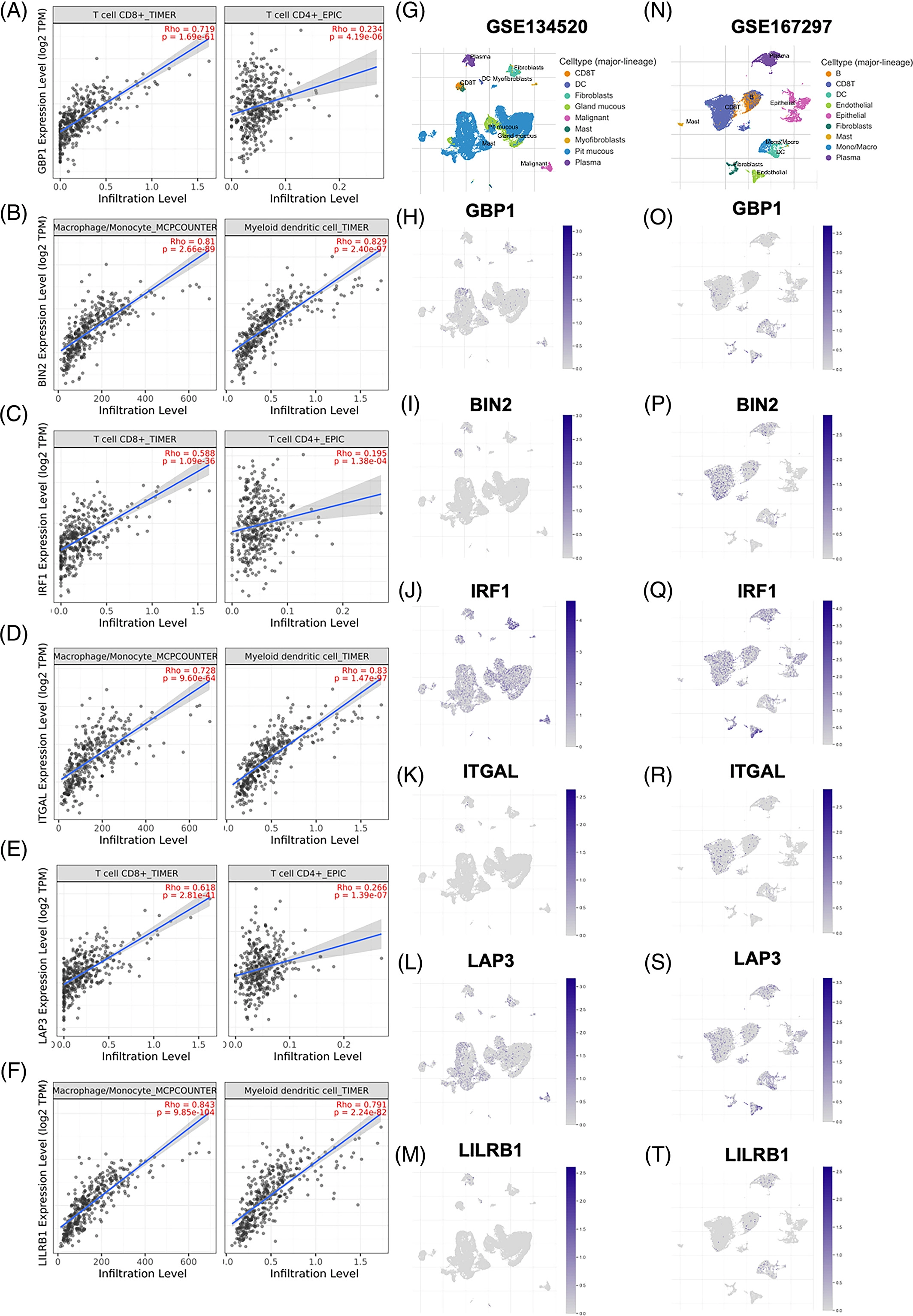
为了进一步证实这些关键的中枢基因与TIICs的特征相关联,应用了TIMER 数据库。GBP1(图5A)、IRF1(图5B)和LAP3(图5C),它们来自蓝色模块,与CD8+T细胞和CD4+T细胞呈正相关。此外,棕色模块基因的高表达,包括BIN2(图5D)、ITGAL(图5E)和LILRB1(图5F),与巨噬细胞/单核细胞和髓系树突状细胞比率的增加有关。为了进一步探索关键HUB基因在GC不同细胞类型中的表达,采用了两个数据集的单细胞测序方法。给出了GSE134520(图5G)和GSE167297(图5N)中GC细胞的情况。GBP1阳性细胞分布于小窝粘液、成纤维细胞、单核/巨噬细胞和胞浆(图5H和O)。BIN2和ITGAL主要表达于CD8+T细胞(图5I、K、P、R)。IRF1在多种免疫细胞和肿瘤细胞中高表达(图5J,Q)。LAP3广泛表达于树突状细胞、内皮细胞、成纤维细胞和恶性细胞(图5L,S)。LILRB1阳性细胞主要与DC相关(图5M,T).
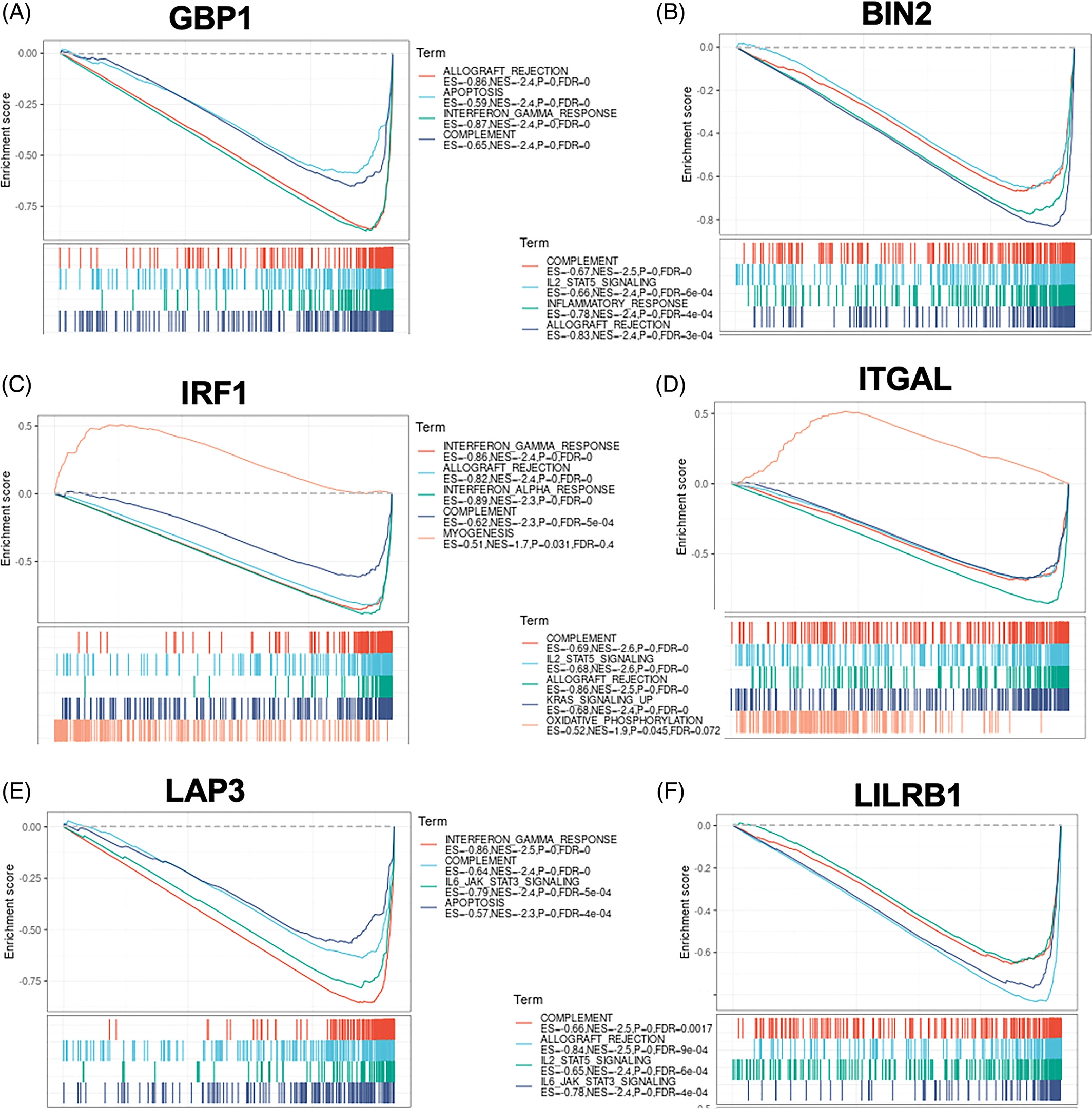
然后,我们利用GSEA对这些关键中枢基因的潜在功能进行了研究。有趣的是,每个蓝色模块基因的高表达都与干扰素-伽马反应呈正相关(图6A、C、E)。此外,较高水平的棕色模块基因与补体系统和IL-2-STAT5信号通路的激活有关(图6B,D,F)。这些结果表明,这些具有代表性的模块基因与GC的免疫调节有关。
图 4:
图 5:
图 6:
图 S4:
图 S5:
表 S7:
表 S8:
小结:
找hub基因:
差异分析方面:用差异分析得到的差异基因和数据库中已知的免疫相关基因取交集,得到了范围更小的十几个基因集。这十几个基因用PPI网络图进行展示。(确认hub基因是差异基因)
WGCNA方面:在蓝色和棕色模块中找到了一百多个hub基因,对这一百多个基因做了GO富集、PPI富集(MCODE插件算法)、KEGG富集。然后从两个模块中各选了前3各基因进行后续分析。(找出hub基因)
hub基因&肿瘤微环境:
上述基因表达量与肿瘤微环境中肿瘤细胞、免疫细胞和基质细胞的相关性,用的是GEO数据库里单细胞测序的数据。
hub基因潜在功能预测:GSEA分析
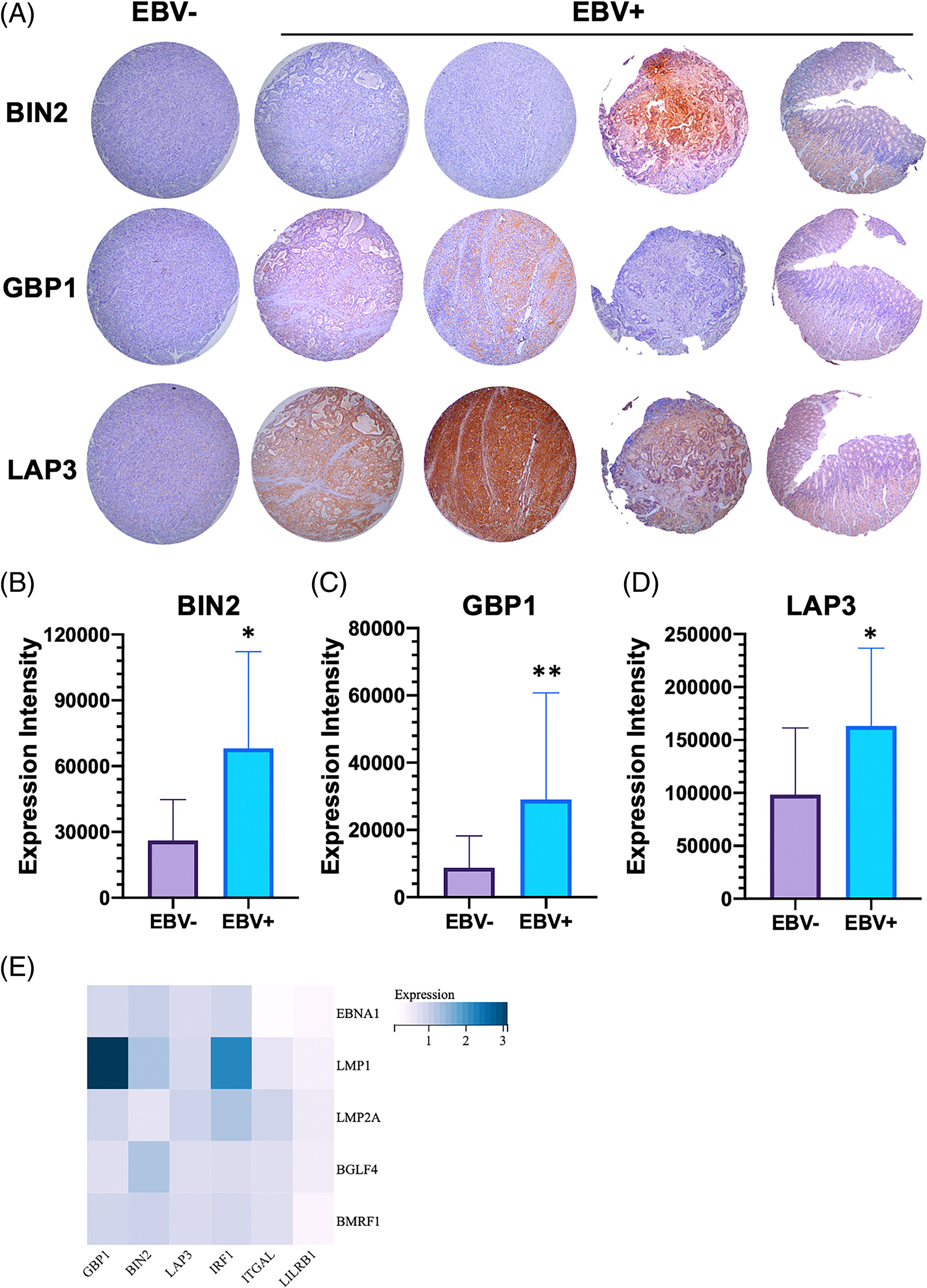
8. EBVaGC组织中GBP1、BIN2和LAP3的高表达
为了检测关键HUB基因在胃癌组织中的表达谱,我们利用免疫组织化学方法研究了GBP1、BIN2和LAP3在胃癌组织芯片中的表达和分布。桂林医科大学共收集并调查了204例胃癌组织。在201例胃癌组织中,EBV阳性19例,占9.45%。如图7A所示,GBP1、BIN2和LAP3主要在胞浆中表达。对组织芯片IHC结果进行组织化学评分,统计结果显示GBP1、BIN2和LAP3在EBV阳性组织中的表达水平高于EBV阴性组织(图7B、C、D)。
9. LMP1过表达调控胃癌关键免疫基因的表达
然后,我们探索了多个EBV病毒基因对这些免疫基因表达的影响。如图7E所示,我们观察到与对照细胞相比,过表达LMP1的MGC-803细胞具有更高水平的GBP1和IRF1,而其他四个基因的表达没有受到显著影响。此外,EBNA1过表达显著降低了ITGAL和LILRB1的表达。需要注意的是,其他几个病毒基因,包括LMP2A、BGLF4和BMRF1,对这些免疫调节分子几乎没有影响。
图 7:
小结:对挑选出的hub基因做了免疫组化来验证,以及病毒相关基因高低表达对hub基因的影响。